






前言
NRF2是细胞应对氧化应激防御的核心转录因子。在健康细胞里,它受KEAP1-CUL3蛋白复合体的严密调控,维持在较低水平。然而,在肺癌、食管癌等多种实体瘤中,NRF2常因基因突变(如NRF2本身的功能获得性突变,或KEAP1、CUL3的功能丧失性突变)而持续激活、不受控制,如同失去刹车的引擎,驱动肿瘤生长并导致其对抗癌治疗产生耐药。
长期以来,科学家们希望找到直接抑制NRF2的药物,但因其属于难以靶向的转录因子而屡屡受挫。于是,研究思路转向了其上游调控枢纽KEAP1-CUL3:能否用小分子重新“激活”这个复合物,恢复其清除NRF2的能力?
近日,拜耳旗下Vividion公司的Matthew P. Patricelli团队及其合作者,研发出一种新型小分子药物VVD-065,该药物是首个通过前所未有的“变构分子胶”机制起效的、可促进NRF2降解的抑制剂。它通过共价结合 KEAP1 的 Cys151 位点诱导构象重排,稳定 KEAP1 与 CUL3 E3 泛素连接酶的相互作用,进而加速 NRF2 的泛素化降解。VVD-065在多种临床前肿瘤模型中展现出显著疗效,目前其相关化合物已经进入 I 期临床试验(NCT05954312),是首个进入临床验证的此类机制药物。
该研究以“A covalent allosteric molecular glue suppresses NRF2-dependent cancer growth”为题,于12月19日在线发表于《Cancer Discovery》上。

研究内容
VVD-065的发现与初步验证
研究团队最初旨在寻找靶向KEAP1 Cys151、并能激活NRF2的小分子(用于自身免疫性疾病的治疗)。然而,在优化先导化合物时,意外发现了一类结构相似的化合物,它们虽然同样共价结合KEAP1的Cys151,却产生了相反的生物学功能:并非稳定NRF2,而是增强KEAP1活性,进而促进NRF2降解。
其中,VVD-065对KEAP1 Cys¹⁵¹表现出纳摩尔级的超高结合效力与卓越的选择性,能有效降低NRF2蛋白水平及其下游靶基因的表达。
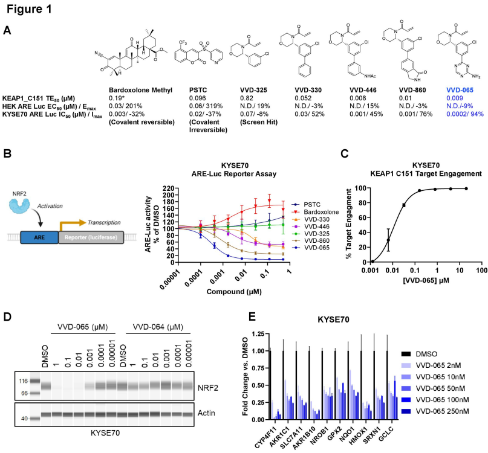
图1. VVD-065 结合 KEAP1 的 Cys¹⁵¹ 诱导 NRF2 降解及靶基因抑制
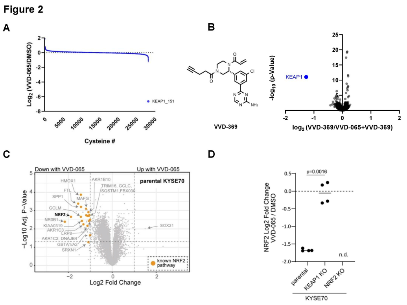
图2. VVD-065 是 KEAP1 的 Cys¹⁵¹ 的高选择性配体
VVD-065 活性的结构基础与机制解析
研究解析了 KEAP1 BTB 结构域与 VVD-065 结合的晶体结构(分辨率 1.87Å,PDB ID: 9DU7),结构显示VVD-065共价结合KEAP1 Cys151,并诱导其BTB结构域发生特异性构象变化(如Cys151旋转、螺旋6向结合口袋位移),整体构象趋向 KEAP1-CUL3 结合态。
生化实验证实了这一观察:HTRF 和SPR实验表明, VVD-065 能够把 KEAP1-CUL3 的结合亲和力从783 nM提升到65nM,解离速率从0.01 s-1 降低到 4.3X10-4 s-1;泛素化实验也进一步证明,VD-065处理增强了KEAP1-CUL3复合物的形成,并加速了NRF2的多聚泛素化过程。
这些证据共同揭示了VVD-065作为“分子胶”的作用本质:通过变构调节,稳定并增强KEAP1与E3连接酶CUL3的相互作用。
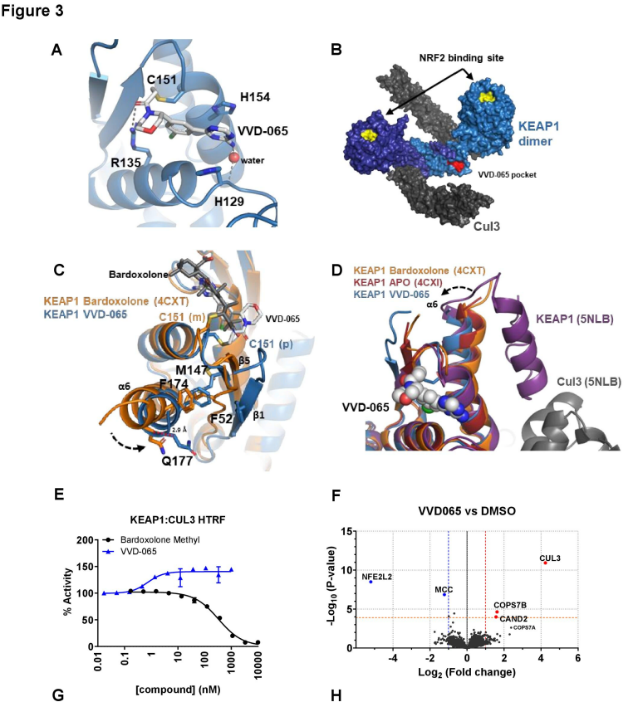
图3(1). VVD-065 稳定 KEAP1-CUL3 复合物形成
细胞水平的功能与依赖性验证
VVD-065 活性依赖 KEAP1-NRF2 相互作用
研究证实,VVD-065促进NRF2降解的功能,严格依赖于KEAP1与NRF2之间残余的结合能力。在KEAP1或NRF2发生特定突变、导致二者结合完全丧失的癌细胞中,VVD-065无效。
VVD-065 的癌细胞生长抑制作用
在三维培养(3D球体)模型中,VVD-065能有效抑制NRF2依赖性癌细胞的增殖,且该抑制效果与化合物降低NRF2蛋白水平的能力高度相关,证实了其作用的靶点特异性。
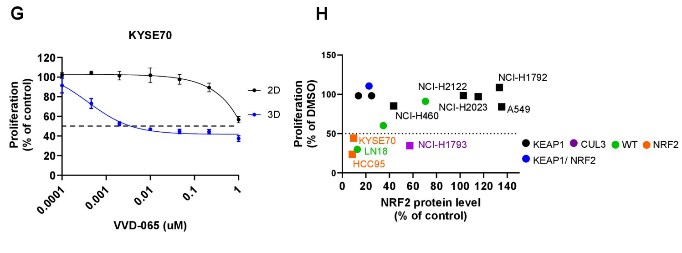
图3(2). 细胞表型
动物模型中的疗效验证
在携带NRF2通路突变肿瘤的小鼠模型中,口服VVD-065能实现良好的肿瘤靶点占据,持续下调NRF2信号,并显著抑制肿瘤生长,此效果在患者来源肿瘤移植模型及免疫系统完整的同源小鼠模型中均得到证实。
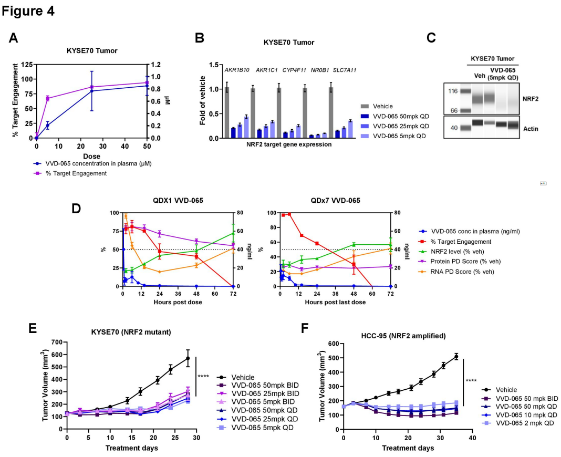
图4. VVD-065 在体内的抗肿瘤效应
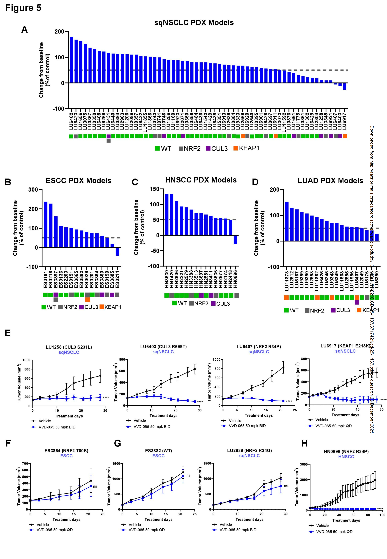
图5. VVD-065 在患者来源异种移植瘤(PDX)模型中的抗肿瘤效应
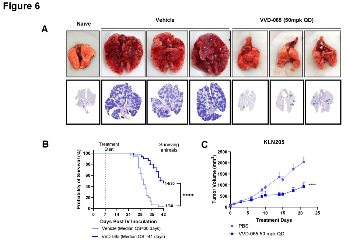
图6. VVD-065在同基因型肿瘤模型中展现出显著的抗肿瘤作用
联合治疗的增效作用
VVD-065与多种标准化疗药物或放疗联合使用时,在临床前模型中显示出强大的协同抗肿瘤效果,能显著逆转由NRF2介导的治疗耐药性。
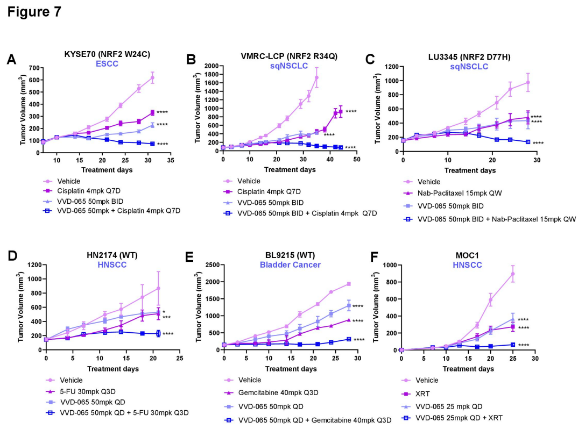
图7. VVD-065与放化疗的协同作用
总结
该研究跳出“直接抑制NRF2”的传统思路,报道了首创的NRF2小分子降解剂VVD-065,它通过一种全新的“变构分子胶”机制发挥作用。该化合物共价结合KEAP1的Cys151位点,诱导其发生构象变化,从而稳定并增强KEAP1与CUL3 E3泛素连接酶的相互作用,进而加速致癌转录因子NRF2的泛素化降解。
这项工作不仅为NRF2持续激活的肿瘤提供了全新的治疗策略,其“通过稳定 E3 连接酶复合物来促进靶蛋白降解”的范式,也为靶向其他传统意义上“不可成药”蛋白开辟了革命性的新路径。
值得一提的是,1.87 Å的高分辨率共晶结构,将“Cys¹⁵¹旋转-螺旋6内移”这一变构开关以及“分子胶”的稳定作用可视化,为后续药物的优化迭代提供了关键结构基础,再次彰显了结构生物学在靶点机制验证与理性药物设计中的不可或缺的作用。












