






前言
G蛋白偶联受体(GPCRs)是一类重要的信号转导蛋白,通常通过激活G蛋白或招募β-arrestin(抑制蛋白)来传递信号。传统观点认为,这两条路径相辅相成,且多数GPCR需要配体激活后才能有效招募β-arrestin。然而,一些“非典型”GPCRs,如GPR1,几乎不激活G蛋白信号通路,但却能招募β-arrestin。
GPR1是由趋化因子样蛋白chemerin激活的受体,参与炎症、脂肪生成和代谢调控。与另一个chemerin受体CMKLR1(典型的GPCR)不同,GPR1能自发(即不依赖激动剂)招募β-arrestin并发生内化,但G蛋白信号极弱。因此,GPR1被认为是一个“诱饵受体”(decoy receptor),通过清除chemerin来调控其信号。
尽管GPR1的这种“诱饵”功能已被提出多年,但其背后的分子机制一直不清楚:它为何能在没有配体的情况下就招募β-arrestin?它为何能同时清除“活性”和“非活性”的chemerin?它又是如何影响细胞的生理功能的?这些问题成为GPCR信号研究领域的重要前沿。
近日,上海药物所/杭高院吴蓓丽团队、临港实验室朱亚团队、上海药物所赵强、谢岑团队和上科大水雯菁团队合作在Science上发表了题为“Noncanonical agonist-dependent and -independent arrestin recruitment of GPR1”的研究文章。
研究通过冷冻电镜首次解析了chemerin 结合态与配体游离态下,GPR1 与两种功能差异显著的 β-抑制蛋白亚型(β-arrestin 1 和 β-arrestin 2)的结合模式,从原子层面阐明了非典型GPCR的独特工作机制,为理解 “诱饵受体” 功能提供了结构依据,也为肥胖等代谢性疾病的靶向研究提供了新方向。
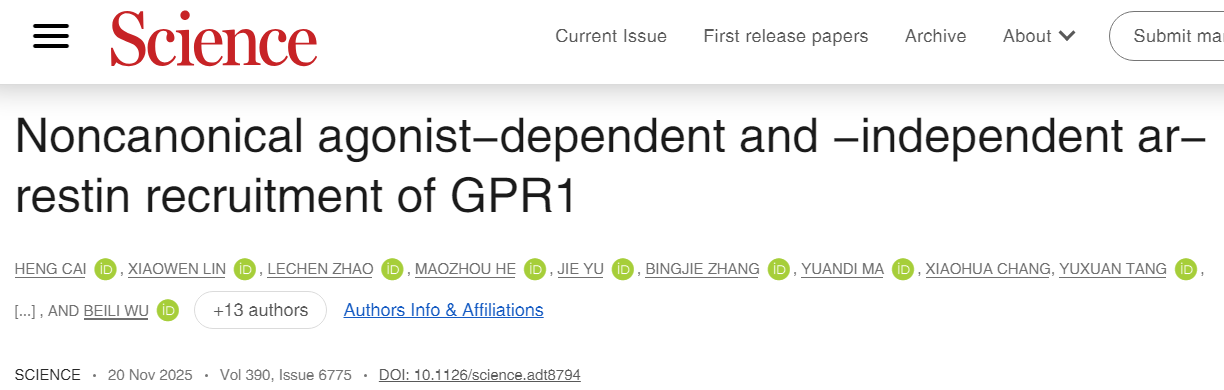
研究内容
1. GPR1与β-arrestin1/2结合模式(配体存在下)
与经典A类GPCR–arrestin复合物中β-抑制蛋白单一结合构象不同,β-arrestin 1 与GPR1结合时有4种不同构象,呈现“动态摆动模型”,从水平到倾斜,可能与内化过程相关。
相比之下,β-arrestin 2以更稳定的方式与GPR1结合,仅有一种结合构象,这可能有助于受体信号传导。
2. 激动剂非依赖性(组成性)arrestin招募
冷冻电镜结构显示在无配体状态下,GPR1处于非激活态,但仍能与β-arrestin1结合,这是由于GPR1的C端组成性磷酸化,形成一个“磷酸化条形码”。内源性脂肪酸(如棕榈油酸和棕榈酸)作为共结合脂质,结合在受体与arrestin的界面,进一步稳定了该复合物。
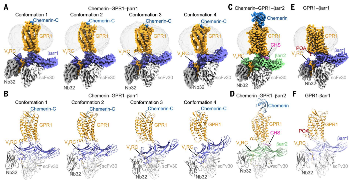
图1. GPR1-βarr复合物的冷冻电镜结构图
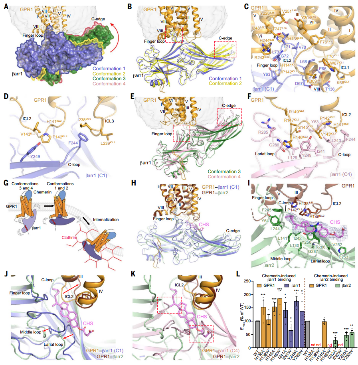
图2. Chemerin诱导GPR1的βarr1和βarr2招募

图3. GPR1 的组成型 arrestin 招募机制
3. 脂质介导的复合物稳定
在无激动剂的GPR1–βarr1结构中,内源性脂肪酸(棕榈油酸和棕榈酸)结合在受体与arrestin的界面,增强相互作用,协助内吞,提示该类脂分子对于 GPR1 清除非激活型chemerin发挥调控作用。
而在chemerin存在的GPR1–βarr2结构中,胆固醇起到类似作用,但只对βarr2有效。

图4. GPR1 与CMKLR1 的 arrestin 内化差异验证
4. GPR1 在脂肪代谢中的功能验证
研究团队进一步在脂肪细胞模型中发现,在高脂环境下,CMKLR1直接促进脂肪代谢,降低脂质积累;而GPR1则通过其高效的“清道夫”功能,持续清除非激活型chemerin,从而间接解放了更多的CMKLR1,使其能够被激动剂型的chemerin激活,最终辅助促进了脂质分解代谢。
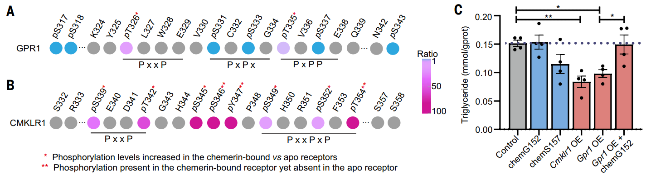
图5. GPR1/CMKLR1 的磷酸化模式与脂质代谢调控
总结
这项研究通过解析GPR1与β-arrestin在不同功能状态下的复合物结构,揭示了其作为一种“诱饵受体”的独特工作机制。研究发现,GPR1打破了经典GPCR的范式:它不仅能在激动剂刺激下,以多种动态构象招募β-arrestin 1驱动高效内化,还能在无激动剂时,借助自身持续的磷酸化及脂肪酸的稳定作用,“自发地”完成arrestin招募。这种独特的组成性活性使其能够持续清除体内的chemerin配体,从而间接解放另一个信号受体CMKLR1,最终在脂肪细胞中促进脂质分解、减少脂质堆积。
该研究首次在原子层面完整描绘了一个非典型GPCR如何通过精巧的结构可塑性与细胞环境中的脂质协同作用,实现其独特的“清道夫”功能。这不仅深化了我们对GPCR信号传导多样性的理解,也为肥胖、脂肪肝等代谢性疾病的靶向治疗提供了新的潜在靶点与理论支撑。












