






前言
CDK4/6抑制剂(如 palbociclib、ribociclib、abemaciclib)联合内分泌治疗已成为HR+/HER2−晚期乳腺癌的标准方案,然而,现有药物对CDK6的同步抑制会带来明显副作用:CDK6在造血干细胞增殖中不可或缺,抑制它会导致程度不一、甚至危及感染的中性粒细胞减少;abemaciclib因额外抑制GSK3β,还伴随胃肠道毒性。临床上不得不通过停药、减量或加用升白针来平衡疗效与安全,既增加负担,也可能削弱对肿瘤的持续压制。
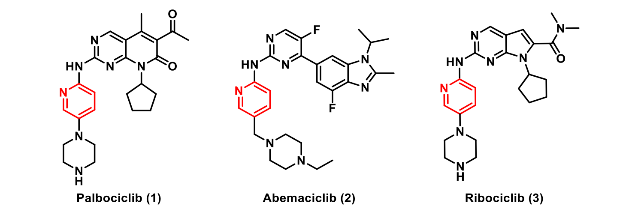
图1. 用于治疗HR+和HER2-乳腺癌的已获批CDK4/6抑制剂
另一方面,基因组学与功能研究表明,乳腺癌细胞周期进展的主要驱动者是CDK4,而CDK6的作用相对次要。基于这一差异,开发一款高选择性CDK4抑制剂,理论上可在保留抗肿瘤活性的同时,减少对骨髓和肠道的连带损伤。
近日,辉瑞研究团队Gary M. Gallego等人通过基于结构的药物设计、分子动力学模拟及脂溶效率导向优化策略,成功开发了一种高选择性CDK4抑制剂Atirmociclib。该抑制剂显著降低了对CDK6的抑制,有望克服现有CDK4/6抑制剂引发的骨髓抑制和中性粒细胞减少等副作用。目前Atirmociclib已在乳腺癌模型中实现肿瘤消退,并进入 III 期临床,有望为 HR+/HER2- 乳腺癌患者提供更安全、更有效的治疗新选择。
研究以“Discovery of Atirmociclib (PF-07220060): A Potent and Selective CDK4 Inhibitor”为题发表在《Journal of Medicinal Chemistry》上。

研究内容
药物发现过程
1. 先导化合物的发现与初步优化
团队通过内部化合物库筛选,确定化合物5为初始优化起点。化合物5虽然具备一定活性(CDK4 Ki = 0.43 nM)和 CDK6 选择性(11倍),但对 CDK2 缺乏选择性(仅5倍),无法满足临床安全性需求。
表1. 筛选其实化合物的初步优化
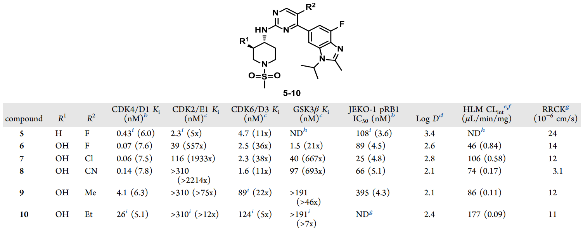
针对化合物5对CDK2缺乏选择性的问题,团队为化合物 5 引入羟基哌啶基团得到化合物 6。该基团与CDK4铰链区His100形成氢键,而CDK2的对应位置是Phe82,无法形成此类相互作用,从而获得557倍的选择性提升。
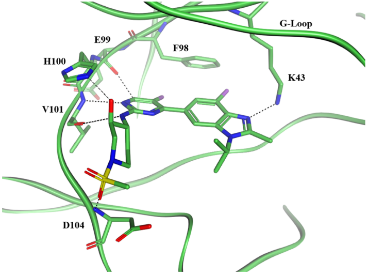
图2. 化合物6在CDK4s蛋白中的共晶结构(2.1 Å,PDB: 9PE7)
针对 GSK3β 抑制引发的胃肠道毒性,团队在嘧啶环 C5 位引入不同取代基。结果显示,氯取代的化合物7 在维持 CDK4 高活性(Ki=0.06 nM)的同时,对 GSK3β 的选择性提升至 667 倍,成为后续优化的先导化合物。
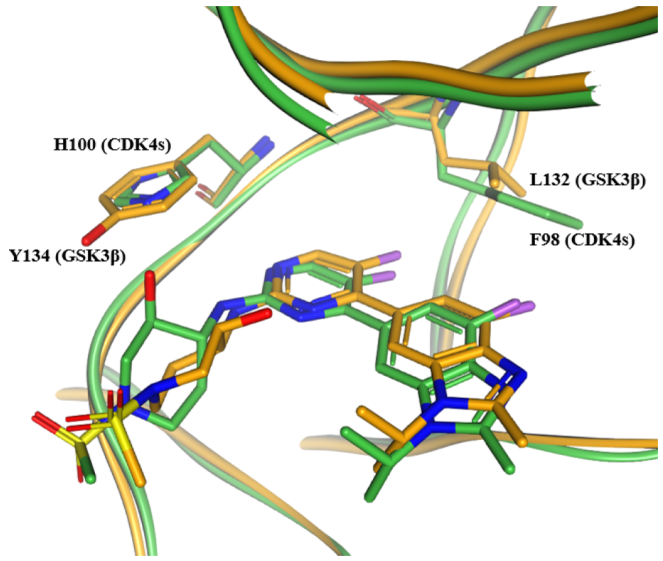
图3. 化合物6在CDK4s(绿色,PDB:9PE7)与GSK3β(橙色,PDB:9PE9)中的共晶结构叠加图
2. 代谢稳定性优化
化合物7虽然活性优异,但存在代谢稳定性问题。代谢研究发现,苯并咪唑2位甲基是主要代谢位点,将其替换为羟甲基得到化合物 11,代谢稳定性显著改善,但其渗透性大幅降低。
表2. 旨在降低代谢的化合物
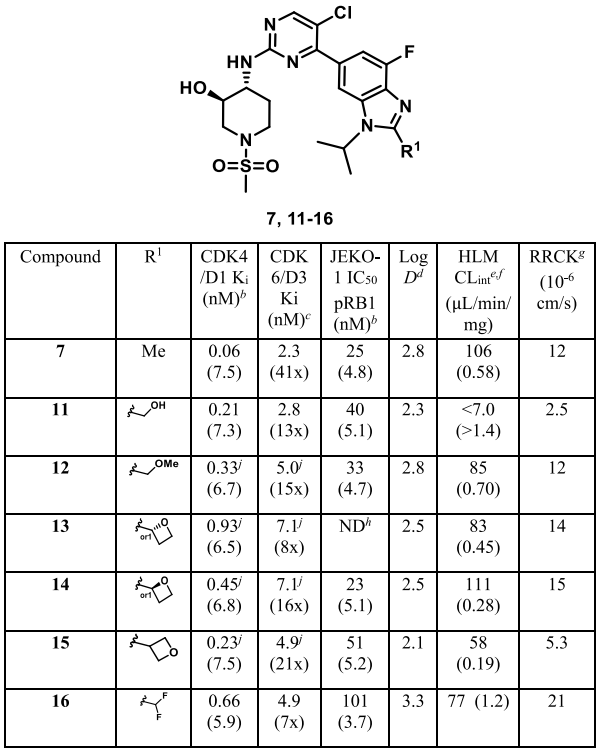
3. 同步优化渗透性与CDK6选择性
团队转而通过降低拓扑极性表面积(TPSA)以提高渗透性,将哌啶环改为四氢吡喃,得到化合物4(即最终的PF-07220060),其渗透性大幅提升,且代谢稳定性良好。
表3. 渗透性和选择性的优化
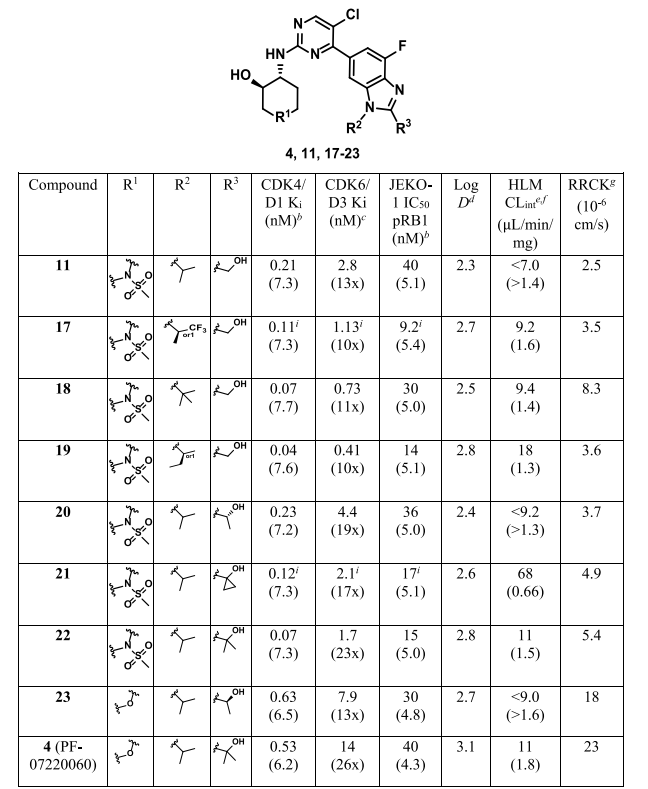
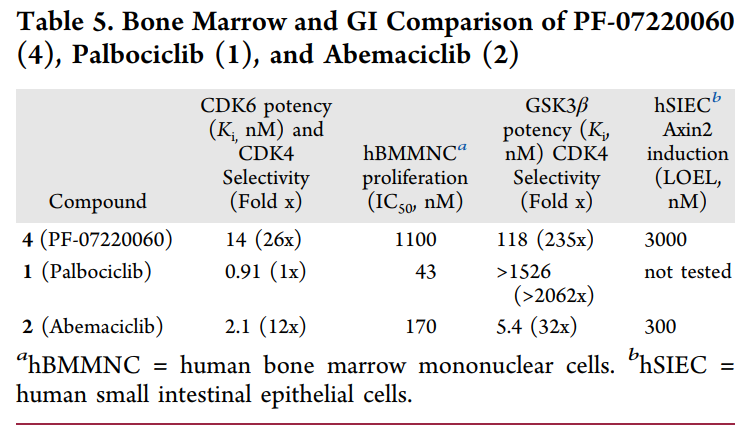
表5. PF-07220060(4)、Palbociclib(1)和Abemaciclib(2)的骨髓和胃肠道比较
分子动力学揭示选择性机制
静态共结晶结构显示Atirmociclib在CDK4s和CDK6中的结合模式相似,但分子动力学模拟揭示了其选择性的根源:CDK4s的G-loop更具构象灵活性,而CDK6的G-loop因存在Glu18、Glu21和Lys26之间的盐桥作用而刚性较强。这种灵活性差异使得CDK4s能更好地容纳化合物4的异丙醇基团,从而产生26倍的CDK4/CDK6选择性。
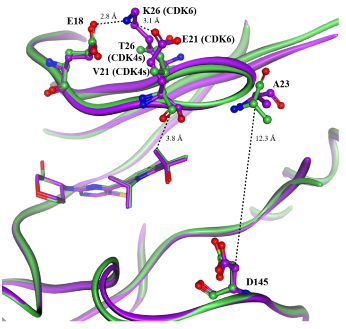
图5. 化合物4(PF-07220060)在CDK4s(绿色,PDB: 9PE8)和CDK6(紫色,PDB: 9D8U)中的共晶结构叠加图
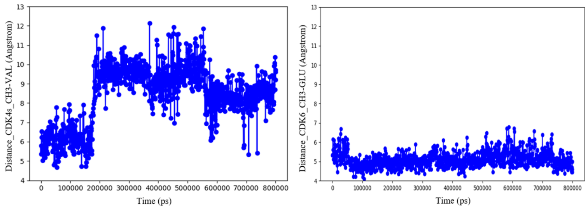
图6. 4(PF-07220060)的甲基与Val21(CDK 4s,左)或Glu21(CDK6,右)之间距离的时间序列
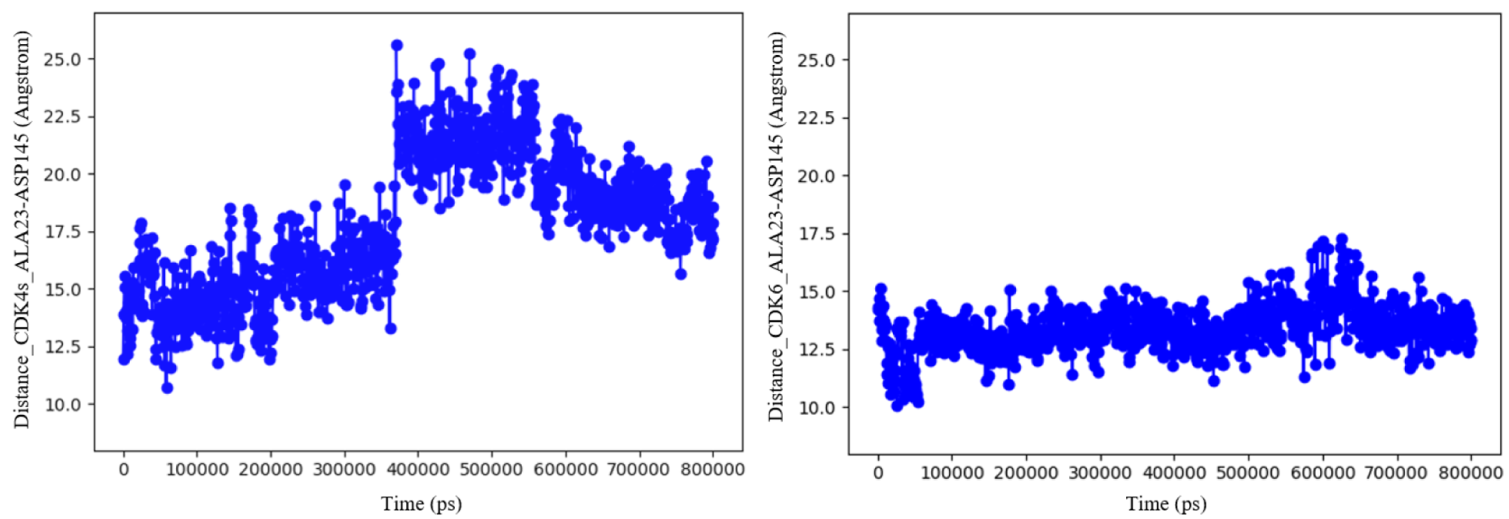
图7. CDK4s Ala23-Asp145(cl.77,左)和CDK6-Ala23-Asp1.45(g.I.9,右)之间距离的时间序列
选择性CDK4抑制剂的体内分析
在ZR75-1(HR+/HER2-乳腺癌)异种移植模型中,Atirmociclib表现出剂量依赖性的肿瘤生长抑制,在高剂量(60 mg/kg,每天两次)下甚至能实现肿瘤消退,且在84天的长期给药中展现出优异的肿瘤控制效果,同时对 pRB 的抑制可维持 24 小时。
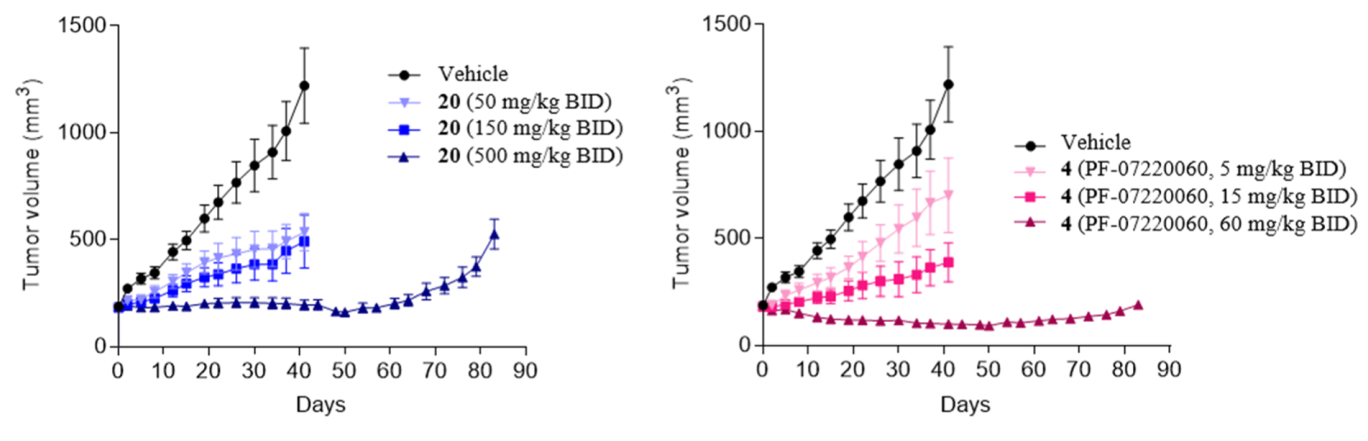
图8. 使用增加剂量的20与4对ZR75-1 HR+ HER2-乳腺癌异种移植物的肿瘤生长抑制
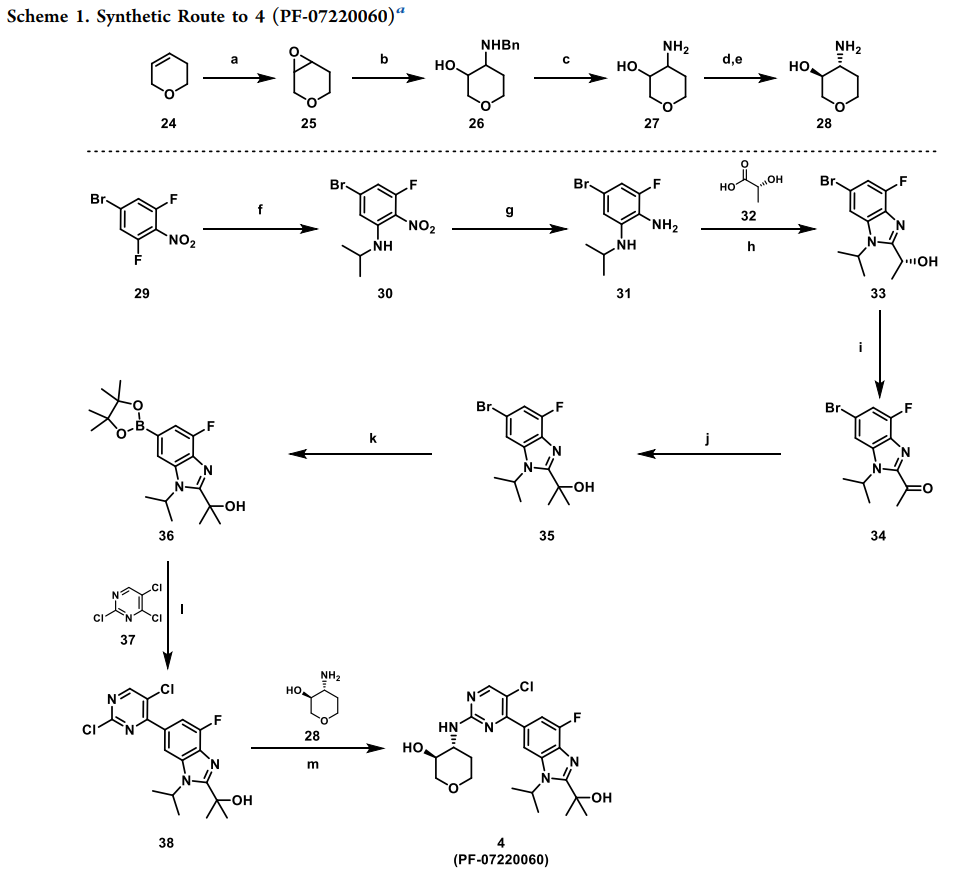
Atirmociclib的合成路线
总结
这项研究成功开发了一种具有新型、强效且高选择性的CDK4抑制剂——Atirmociclib(PF-07220060)。其核心成果在于,通过结构导向的药物设计与多参数理性优化相结合的策略,在保留卓越抗肿瘤活性的同时,显著降低了对CDK6和GSK3β的抑制,从而在临床前模型中展现出更优的安全性潜力。
临床层面,Atirmociclib为解决现有CDK4/6抑制剂常见的骨髓抑制和胃肠道毒性提供了新路径,有望为乳腺癌患者带来疗效相当但耐受性更佳的治疗选择。药物研发层面,它成功示范了如何利用亲脂性效率、分子动力学模拟与结构生物学等工具,攻克针对高度同源靶点的选择性难题,为开发下一代精准靶向药物提供了宝贵范式。目前,该候选药物已进入后期临床试验阶段,其进展值得期待。











