






前言
轴突退行性病变是多种神经系统疾病的共同病理特征,SARM1作为调控轴突退化的核心NAD + 水解酶,正常生理状态下其处于自抑制构象;但是当细胞受损时,NAD+ 水平下降、烟酰胺单核苷酸(NMN)累积,会触发 SARM1 激活并快速降解 NAD+,最终引发轴突退行性病变和细胞死亡。因此,抑制SARM1被认为是治疗多种神经退行性疾病的潜在策略。
目前已报道的SARM1抑制剂主要为碱基交换抑制剂(BEIs),这类分子利用反应性杂环弹头与 NAD + 反应,从而阻断酶促反应并发挥轴突保护作用。但Genentech、Roche等多个团队发现BEIs 在亚抑制浓度下会反常激活 SARM1,反而加重神经损伤。薛定谔公司联合全球顶尖创新药企百时美施贵宝(BMS)针对这个靶点做了相关研究,以期利用计算化学驱动SARM1抑制剂的开发。
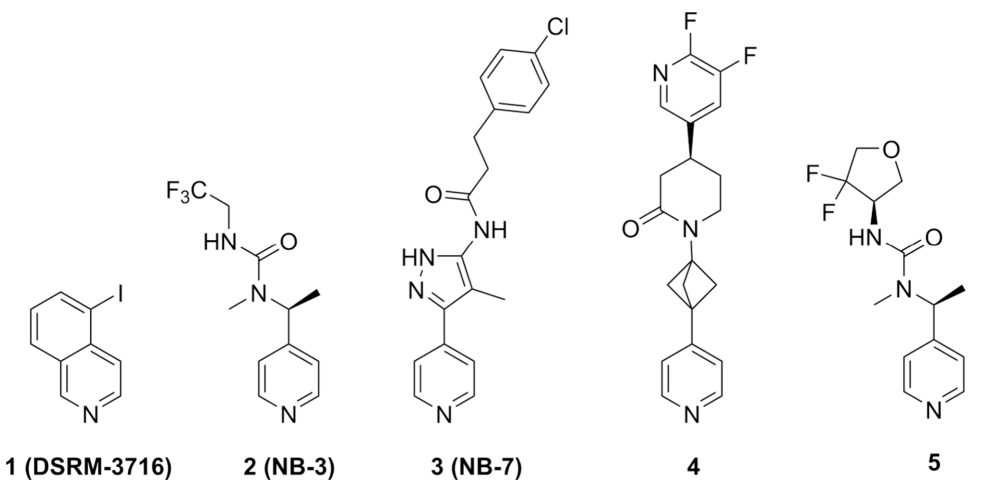
图1. 已报道的含反应性弹头的SARM1抑制剂
薛定谔公司Adam M. Levinson等人报道了一系列咪唑并[4,5-c]吡啶类SARM1抑制剂的发现及优化过程,核心成果包括:
方法创新:次针对SARM1碱基交换抑制剂建立了自由能微扰(FEP+)计算流程,解决了前药与活性加合物结构不一致的模拟瓶颈;
结构新发现:首次揭示了W662一种新的配体诱导旋转异构状态,拓展了SARM1结合口袋的可药性空间
分子创新:开发了咪唑并[4,5-c]吡啶类新型SARM1抑制剂骨架,获得纳摩尔级工具分子;
安全警示:明确低剂量反常激活是 BEIs 类抑制剂的共性风险,提示临床开发必须保证高且持续的靶标结合与药物暴露,避免亚抑制浓度引发反常激活
相关研究以“Structure-Based Discovery of Imidazo[4,5-c]pyridine SARM1 Modulators Showing Paradoxical Activation”为题,4月8日在线发表于《Journal of Medicinal Chemistry》。
研究内容
1. 新型 BEI 设计与建模流程
BEI 并不是以其原始结构结合靶点,而是先与 NAD⁺ 反应形成一个共价加合物,该加合物才是真正结合在活性位点的分子。常规的 FEP+ 无法直接处理这种“反应后”的配体。
因此,团队创新性的采用先虚拟构建药物 NAD⁺ 共价加合物,再通过FEP+预测其结合亲和力的建模流程,最终成功预测了新型双环支架(如咪唑并吡啶类)的结合模式,加速了苗头化合物的发现。
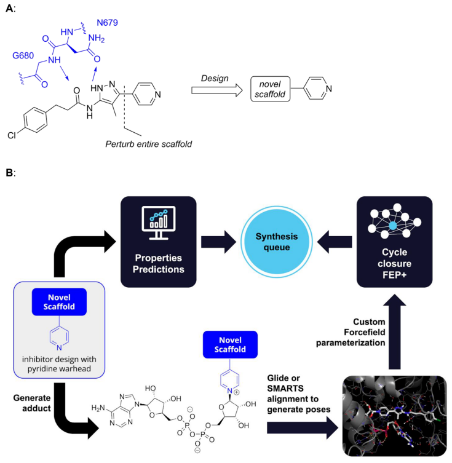
图2. 用于发现 SARM1 抑制剂的设计思路与FEP +建模流程
2. 杂双环BEI 骨架的发现
利用上述流程,团队以咪唑并[4,5-c]吡啶双环骨架替换原有的吡唑酰胺,获得了溶解度与活性更优的化合物7,为后续优化奠定了全新化学起点。
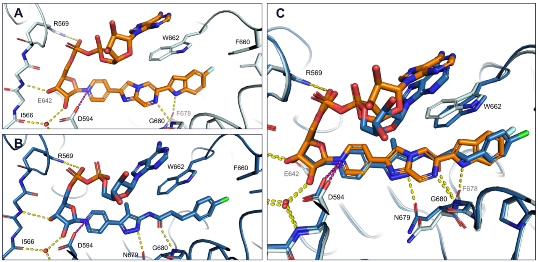
图3. 化合物6与SARM1的TIR结构域复合物晶体结构
3. 结构指导的 SAR 优化
以新骨架为起点进行侧链优化,通过晶体结构解析,团队发现乙二胺侧链诱导 W662 旋转约 90°,形成全新可药结合口袋,基于此优化最终获得了纳摩尔级活性分子19。
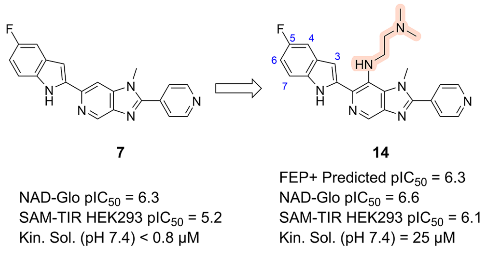
图4. 相对于初始苗头化合物7,发现一个可改善细胞活性和动力学溶解度的新取代位点
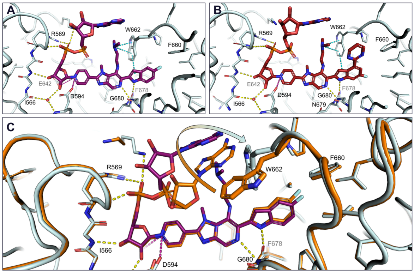
图5. 代表性抑制剂与SARM1的共晶结构
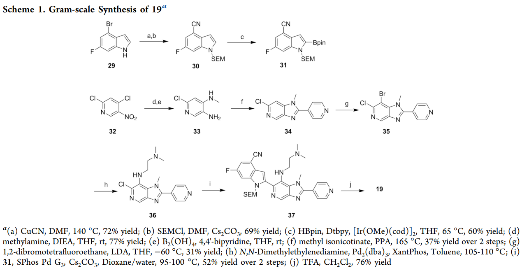
路线1. 化合物19的克级制备路线
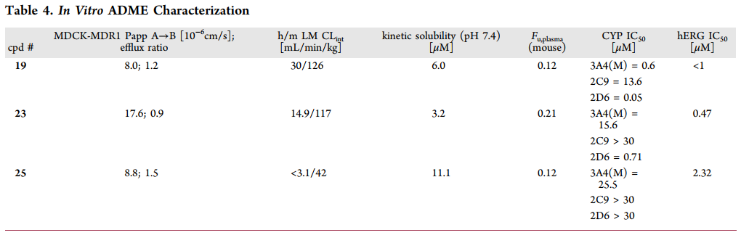
4. ADME与安全性优化
针对19的CYP抑制与hERG毒性问题,团队将吡啶弹头改为哒嗪、碱性胺改为中性氧杂环丁烷,得到代谢稳定性、口服生物利用度及中枢渗透能力更佳的优化分子25。
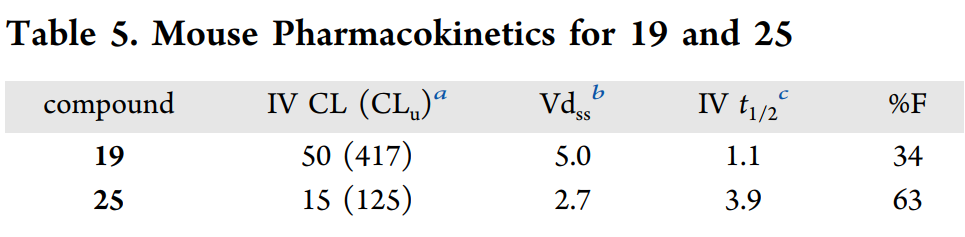
5. 低剂量反常激活效应的发现
在小鼠神经损伤模型中,低剂量19反而加重了神经损伤标志物;进一步实验证实,在轻度应激下,亚抑制浓度的碱基交换抑制剂会异常激活SARM1,加剧细胞死亡。这提示此类药物必须在高剂量、充分靶标结合的情况下才能发挥治疗效果。
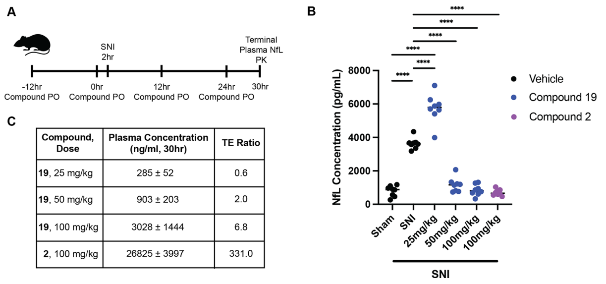
图6. 小鼠SNI 模型中化合物19 的体内疗效与低剂量反常效应
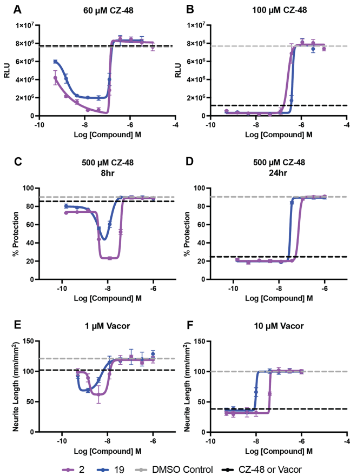
图7. 体外低浓度BEI 反常激活SARM1 并加剧神经退变
总结
本研究成功开发了一类结构新颖的咪唑并 [4,5-c] 吡啶类 SARM1 抑制剂,通过计算辅助设计与结构导向优化,获得了兼具高活性、长滞留时间与一定中枢渗透能力的工具分子,并首次揭示了配体诱导 SARM1 关键残基 W662 发生特征性旋转的结合模式,为 SARM1 调节剂的理性设计提供了全新思路与结构基础。
同时,研究明确了BEIs存在低剂量反常激活的共性风险,提示临床开发必须保证充分且持续的靶标暴露,对提升 SARM1 靶向药物研发的安全性与有效性具有重要指导意义。












