






前言
大麻的医学应用历史跨越千年,唐代《本草拾遗》和古罗马《药物论》中,就已经记载了它具有镇痛与情绪调节的价值;现代研究进一步证实,大麻缓解疼痛、改善焦虑抑郁的效应主要源于其对内源性大麻素系统的调控。
在这一系统中,大麻素受体1(CB1)是中枢神经系统中表达最丰富的G蛋白偶联受体之一,是介导上述治疗效应的核心分子开关。CB1受体下游主要包含两条关键信号通路:Gi蛋白通路(主要负责镇痛、抗焦虑等治疗效应)和β-arrestin通路(与镇痛耐受、成瘾等副作用密切相关)。
传统CB1激动剂在激活Gi蛋白通路产生镇痛效应的同时,也会招募β-arrestin,从而引发一系列严重的副作用,限制了其临床应用。如何在保留其治疗效应的情况下规避副作用,实现“去毒留效”,一直是神经科学和药物研发领域的核心难题。
近日,浙江大学李晓明、董晓武、张岩等研究人员通过结构引导的合理设计,成功开发出两种Gi偏向性CB1激动剂,在保留强效镇痛作用的同时,显著减少了传统CB1激动剂的成瘾与耐受等副作用。
这项研究标志着非阿片类镇痛药物研发迈出了关键一步。研究成果以“Rational design of Gi-biased CB1 agonist with reduced side effects”为题,4月13日在线发表于《Cell》上。

研究内容
1. 发现决定信号偏向性的关键氨基酸残基
在团队过往的研究中,解析了CB1受体与不同信号蛋白复合物的高分辨率冷冻电镜结构,本次研究通过对比发现,在β-arrestin结合状态下,配体结合口袋中的三个氨基酸残基(F189、F268、F379)与激动剂FUB产生明显的空间位阻。这种位阻是驱动FUB采取更深插入构象、进而有效招募β-arrestin的关键。
进一步的功能突变实验证实,将这些残基突变为丙氨酸后,β-arrestin信号显著下降,而Gi信号基本不受影响。这说明这三个残基是调控β-arrestin通路的关键位点。
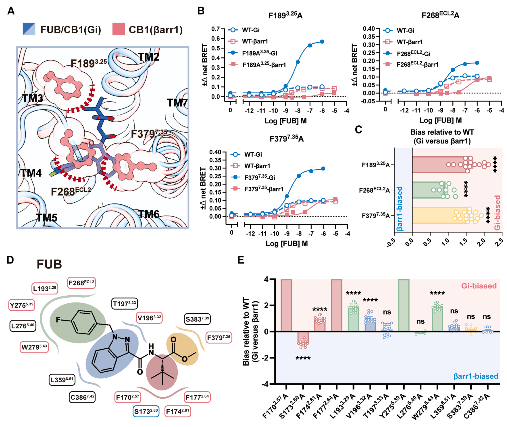
图1.1 发现决定偏向性的关键残基
2. 理性设计Gi偏向激动剂
基于上述冷冻电镜结构,团队对传统的大麻素激动剂 FUB 进行改造:把柔性叔丁基替换成空间更小、更刚性的环丙烷(LZD503)或1-羟乙基(LZD505),成功开发出高选择性Gi偏向激动剂LZD503、LZD505。
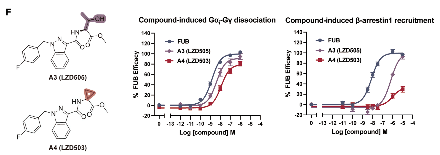
图1.2 Gi偏向型CB1激动剂的设计
3. 冷冻电镜结构验证
为了验证设计策略,团队分别解析了LZD503和LZD505与CB1-Gi蛋白复合物的高分辨率(2.4 Å)冷冻电镜结构。结果显示,这两种激动剂在Gi结合状态下的插入深度与FUB相似;但在β-arrestin结合状态时,由于空间位阻减小,它们无法像FUB那样深埋入结合口袋。这种“浅结合”构象直接解释了它们为何无法有效招募β-arrestin,从而实现了信号偏向。
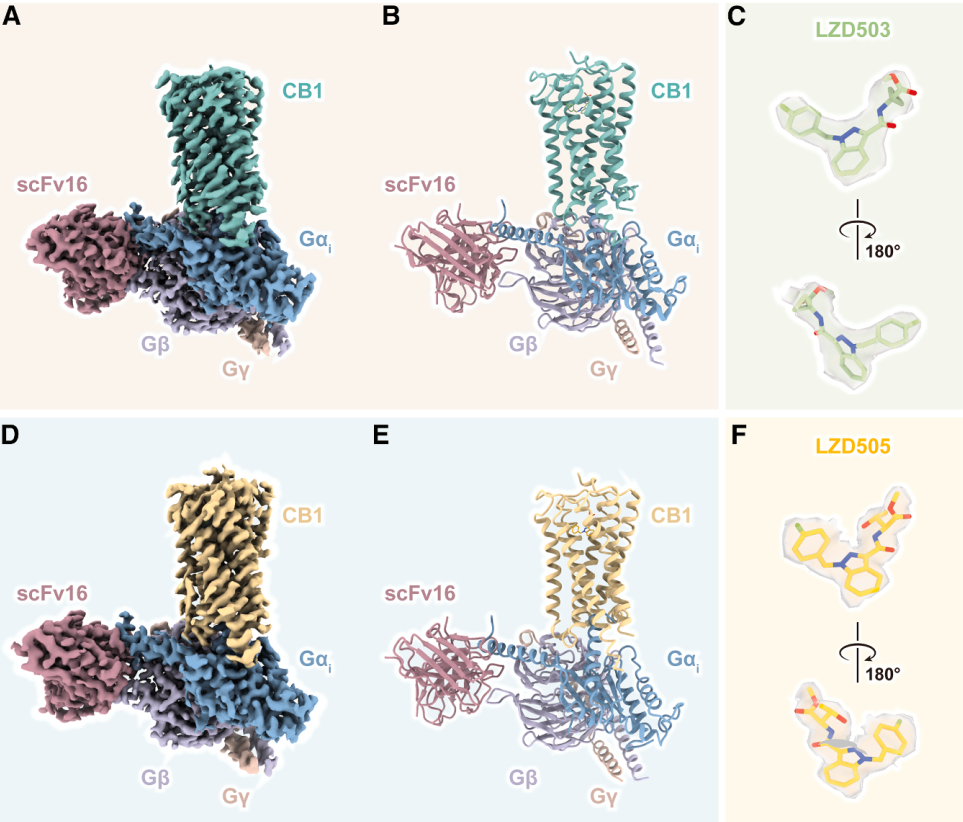
图2. LZD503和LZD505结合的CB1-Gi复合物冷冻电镜结构
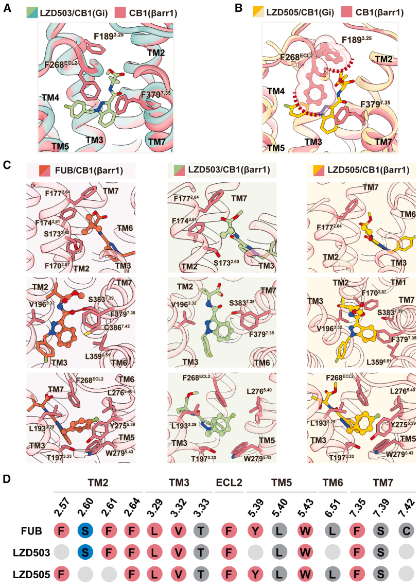
图3. 配体结合口袋决定β-arrestin 信号的结构基础
4. 药效与安全性评估
药效评估:体内动物实验结果表明,LZD503和LZD505在热板、福尔马林、CFA 炎症、SNI 神经病理性疼痛模型中,均能显著缓解疼痛,且可被CB1拮抗剂阻断。
安全性评估:连续7天给药后,LZD503 几乎无镇痛耐受,LZD505 也仅表现出轻微的耐受;与FUB相比,它们引起的体温下降、运动抑制显著减轻;条件位置偏好实验显示无成瘾性。
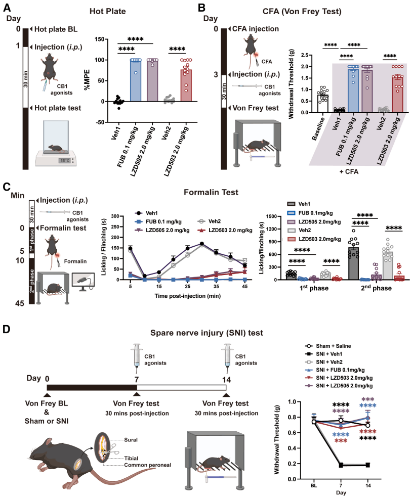
图4. 设计的CB1激动剂在不同小鼠疼痛模型中的镇痛作用
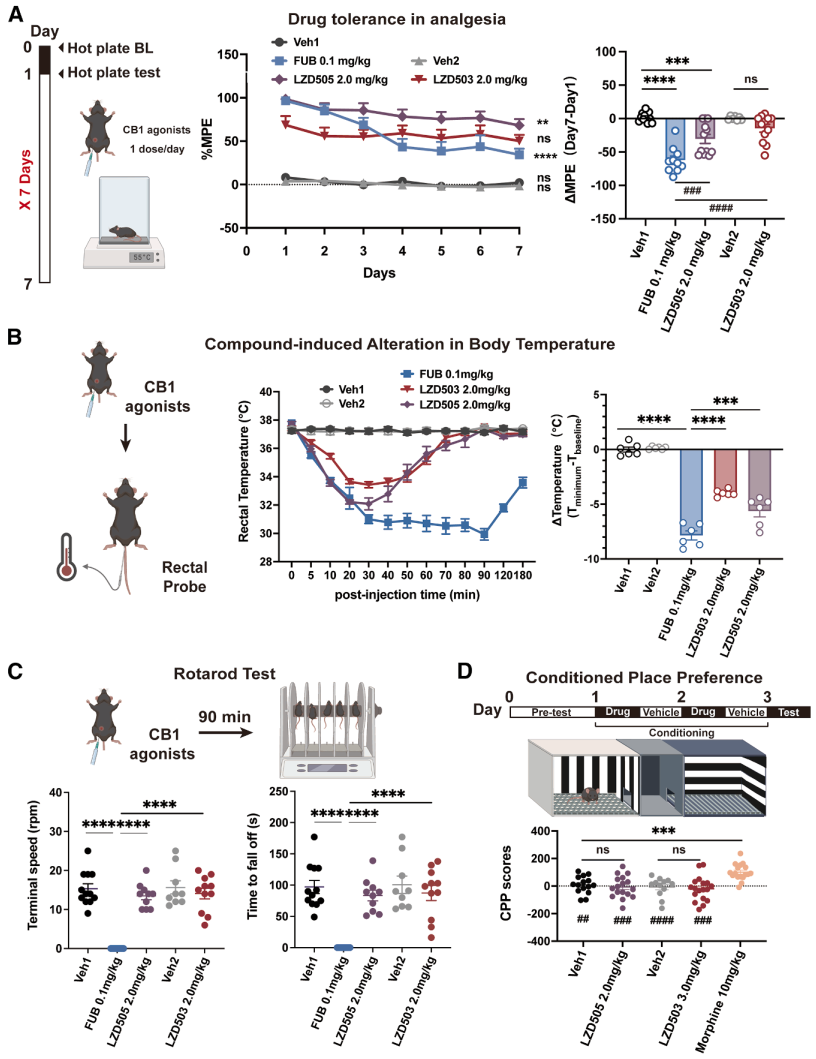
图5. 设计的CB1激动剂LZD503和LZD505的安全性比较评估
总结
这项工作通过结构生物学驱动的理性设计,成功开发出两种Gi偏向性CB1激动剂,在保留强效镇痛作用的同时,显著减少了传统CB1激动剂的多种副作用。不仅为非阿片类镇痛药的开发提供了新方向,也为GPCR偏向性药物设计提供了可推广的结构策略。
从古代典籍中的经验记载,到结构生物学的精确解析,千年之后,这项研究终于为“大麻是药是毒”的古老疑问给出了一个清晰的科学答案:Gi通路留效,β-arrestin去毒,大麻类药物有望真正走出争议,成为安全、可控的现代镇痛工具。











