基于结构的一站式技术服务平台
青云瑞晶关键核心技术
突破纳米晶体结构解析瓶颈,探索微晶结构新世界
精确解析药物分子结构,科学筛选优势晶型
以固态技术提升药物开发成功率
无需结晶,解析高分辨率大分子结构,突破传统结构解析瓶颈
大分子结构解析|冷冻电镜数据处理|纳米颗粒高分辨率表征


实时了解青云瑞晶最新资讯及行业热点新闻

在细胞中,有一类能在细胞膜内部直接切割蛋白质的酶,称为膜内蛋白酶(i-CLiPs)。它们参与调控许多关键的细胞过程,i-CLiPs根据催化机制可分为四类,其中,天冬氨酸蛋白酶家族包含早老素(PS)家族和信号肽肽酶(SPP)家族。
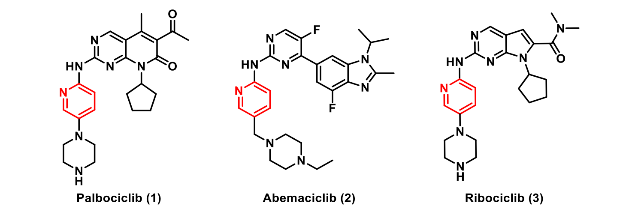
CDK4/6抑制剂(如 palbociclib、ribociclib、abemaciclib)联合内分泌治疗已成为HR+/HER2−晚期乳腺癌的标准方案,然而,现有药物对CDK6的同步抑制会带来明显副作用:CDK6在造血干细胞增殖中不可或缺,抑制它会导致程度不一、甚至危及感染的中性粒细胞减少;abemaciclib因额外抑制GSK3β,还伴随胃肠道毒性。
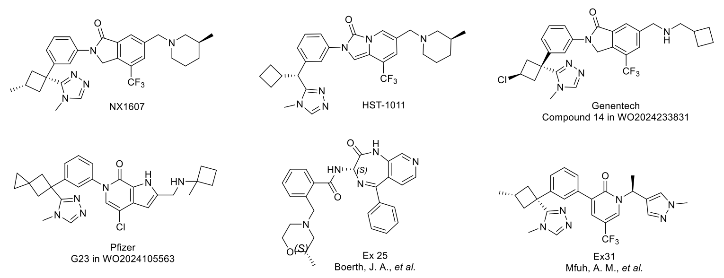
在癌症治疗领域,免疫疗法通过激活患者自身免疫系统攻击肿瘤,已成为一种革命性策略。
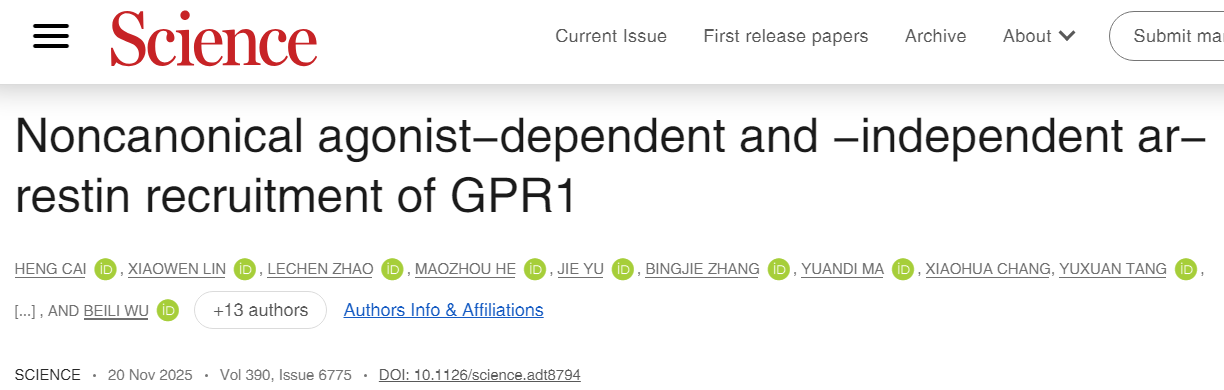
G蛋白偶联受体(GPCRs)是一类重要的信号转导蛋白,通常通过激活G蛋白或招募β-arrestin(抑制蛋白)来传递信号。

+86 18962587269(业务咨询-微信同号)
+86 15606230810(求职招聘-微信同号)

江苏省苏州市常熟高新技术产业 开发区贤士路 88 号 6 幢1001
广东省深圳市宝安区西乡街道共乐社区铁仔路52升业空间A栋317

bussiness@readcrystal.com(业务报价)
contact@readcrystal.com(其他合作)
hr@readcrystal.com(求职招聘)






版权所有 © Copyright 2024 苏州青云瑞晶生物科技有限公司 苏ICP备2021032617号 苏公网安备32058102002758号